Cuando diagnostican a algún familiar o amistad cercana de una enfermedad neurodegenerativa, el mundo se detiene. Sobre todo, cuando comprendemos en profundidad lo que significa la palabra neurodegenerativa: es una enfermedad que afecta al sistema nervioso (al cerebro, médula espinal y/o nervios) y que empeora con el tiempo. Y suelen surgir en pacientes y familiares dos preguntas: ¿cómo va a evolucionar la enfermedad? y ¿cuánto se puede vivir con esto?
Y, aunque para responderlas, los y las especialistas empleen las estadísticas que recogen datos de pacientes descritos en la literatura, todos y todas conocemos casos de personajes públicos que tras ser diagnosticados con una enfermedad neurológica degenerativa, han vivido varios años más de la media, como es el caso del actor Michael J. Fox, con más de treinta años de evolución de la enfermedad de Parkinson, o del científico Stephen Hawking, que convivió cincuenta y cinco años con la esclerosis lateral amiotrófica (ELA). Desgraciadamente, en el caso de las enfermedades priónicas (de las que conocemos como representante a la enfermedad de las vacas locas) estos tiempos de supervivencia son mucho más breves.
En humanos, la enfermedad priónica más frecuente es la enfermedad de Creutzfeldt-Jakob esporádica. Es una enfermedad rara, ya que afecta a un caso entre un millón, por lo que cada nuevo paciente se aleja mucho de “ser un caso más”, siendo muy importante no solo acompañar al paciente y a la familia desde el diagnóstico, sino que estudiar en profundidad a cada paciente tiene gran relevancia.
Se trata de una enfermedad de declaración obligatoria desde hace casi treinta años, por lo que los especialistas están obligados a notificar a Salud Pública cada nuevo paciente diagnosticado. Esto ha permitido disponer de una información exhaustiva de la gran mayoría de los casos que existen, no solo a nivel comunitario o estatal, sino a nivel mundial, y, así, poder devolver a las familias las respuestas a esas preguntas que nos plantean. En España, se sabe que la supervivencia media descrita es de cinco meses y que el 90 % de los casos fallecen a lo largo del primer año, similar a lo descrito en poblaciones como EEUU, pero circunstancias que no se repiten en poblaciones como Japón o Taiwán, donde triplican esta supervivencia.
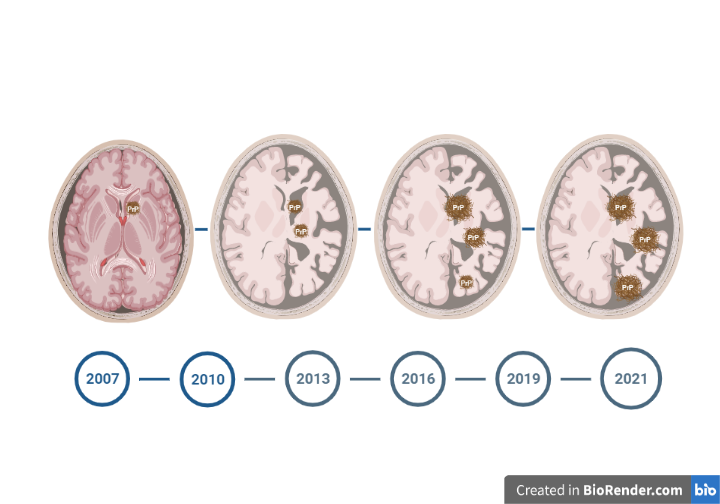
En el País Vasco, y concretamente en Álava, hay una mayor incidencia de estas enfermedades priónicas, motivo por el que en el Hospital Universitario de Araba se encuentra el nodo de referencia del Biobanco Vasco de cerebros con esta patología. Y es en este entorno donde surge el trabajo colaborativo de investigadores e investigadoras del Instituto de Investigación Sanitaria Bioaraba con investigadores del Prion Reseach Lab del CIC Biogune, en el que nos muestran que cada caso, cada persona, es única1. Y en este artículo lo es por dos motivos. El primero, porque la paciente vivió 14 años tras la aparición de las primeras manifestaciones compatibles con la enfermedad de Creutzfeldt-Jakob esporádica, muy superior a los cinco meses de media. Y el segundo, porque como enfermedad rara y poco conocida que es, el diagnóstico en vida de esta paciente fue otro, y fue el estudio de autopsia el que determinó el correcto diagnóstico de la enfermedad que nos ocupa.
Por ello, la Dra. Izaro Kortazar, miembro del equipo investigador, destaca la importancia de realizar la autopsia en este tipo de casos inusuales o como prueba confirmatoria del diagnóstico en casos con sospecha de enfermedad priónica, “A sabiendas que la necropsia conlleva que, en momentos sumamente delicados, a cada familia le toca tomar una decisión aún más delicada y plenamente generosa hacia la comunidad científica, aportando su granito hacia un futuro distinto”.